
Abstract
The U.S. Food and Drug Administration (FDA) evolved from a weak bureau into a globally respected regulator only after major preventable disasters forced legal and cultural change. The Central Drugs Standard Control Organisation (CDCSO) of India now faces a similar inflection point following the deaths of more than 300 children linked to contaminated cough syrups exported from India. This article analyzes the FDA’s historical transformation—focusing on the Elixir Sulfanilamide (1937) and thalidomide (early 1960s) crises—and extracts actionable lessons for the CDCSO. It argues that without structural centralization, mandatory raw‑material testing, transparent recall systems, and a cultural shift toward patient‑first enforcement, India’s “pharmacy of the world” status will remain fragile.
1. Introduction
The transition of the U.S. Food and Drug Administration from a small chemistry laboratory into a globally influential regulator did not happen by design. It happened through catastrophe. Today, the Central Drugs Standard Control Organisation (CDCSO) stands at a similar crossroads. Following international outrage over contaminated paediatric cough syrups that caused child deaths in The Gambia, Uzbekistan, and Cameroon (2022–2023), India’s reputation as the “pharmacy of the world” now depends on whether the CDCSO can pivot from a volume‑driven facilitator to a quality‑obsessed enforcer.
2. The FDA Blueprint: Reform Born of Tragedy
The modern FDA was forged in two distinct medical disasters.
2.1 Elixir Sulfanilamide (1937)
In 1937, a U.S. company dissolved the antibacterial drug sulfanilamide in diethylene glycol (DEG)—a cheap industrial solvent and antifreeze component. The manufacturer conducted no toxicity testing. Within months, 107 people, many of them children, died from DEG poisoning (Wax, 1995). The same toxin was later found in Indian cough syrups exported to low‑income countries.
The public outcry forced the U.S. Congress to pass the 1938 Food, Drug, and Cosmetic Act, which ended the era of “marketing first, testing later.” For the first time, manufacturers had to demonstrate safety before a drug could be sold (FDA, 2022).

2.2 Thalidomide (1960–1962)
Thalidomide was marketed globally as a safe sedative for morning sickness. It caused an estimated 10,000 children to be born with severe limb deformities (phocomelia) worldwide (Vargesson, 2015).
The United States largely escaped this tragedy because of one person: Dr Frances Oldham Kelsey, an FDA reviewer who refused to approve thalidomide despite intense pressure from the company, citing insufficient safety data (Bren, 2015).
The Kefauver‑Harris Amendment (1962) followed. It required drug manufacturers to prove efficacy—not just safety—and mandated informed consent for clinical trial participants (FDA, 2022).
Key lesson for India: Regulatory courage at the individual reviewer level, backed by legal authority, saves lives.
3. The CDCSO Crisis: Responding to the Contaminated Syrup Deaths
Between 2022 and 2023, the World Health Organization (WHO) issued medical product alerts linking Indian‑manufactured cough syrups to acute kidney injuries and child deaths in The Gambia, Uzbekistan, and Cameroon (WHO, 2022; WHO, 2023). Laboratory analyses confirmed unacceptable levels of diethylene glycol (DEG) and ethylene glycol (EG).
Indian government investigations found that certain manufacturers had not performed batch‑wise testing of incoming raw materials, specifically excipients such as propylene glycol and glycerin (Ministry of Health & Family Welfare, 2023).

4. Required Transformations for the CDCSO
To regain international trust, the CDCSO must move beyond reactive circulars. Four structural shifts are necessary.
4.1 Unified Federal Oversight
India currently operates 36 state‑level drug regulatory authorities with no mandatory uniformity. This fragmented system allows non‑compliant firms to engage in “forum shopping”—moving production or licenses to states with weaker enforcement.
Recommendation: Establish a single, autonomous central drug authority with direct enforcement powers under the Drugs and Cosmetics Act, 1940. A unified inspectorate would end the current regulatory patchwork.
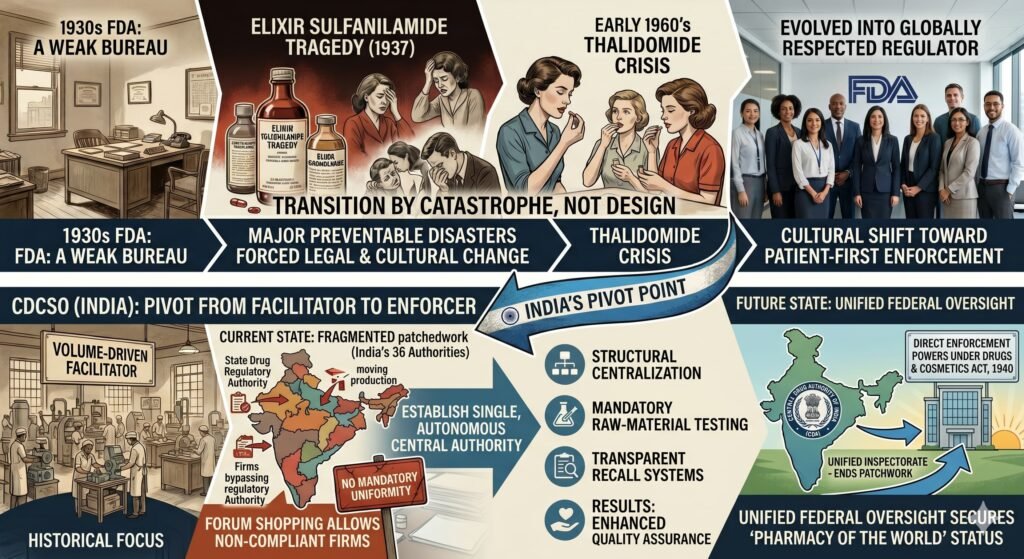
4.2 Mandatory, Verifiable Raw Material Testing
In 2023, the CDCSO mandated that manufacturers test every batch of excipients and active pharmaceutical ingredients (APIs). However, without a centralized digital tracking system, this mandate remains largely honor‑based.
Recommendation: Create a national, real‑time supply chain database. Any batch of propylene glycol or glycerin used in paediatric liquid formulations must be traced from refinery to finished product, with test certificates uploaded before manufacturing begins.
4.3 Strict Enforcement of Revised Schedule M (GMP)
India revised Schedule M of the Drugs and Cosmetics Rules (Good Manufacturing Practices) to align with WHO standards in 2023. Enforcement, however, lags. A risk‑based inspection model is needed.
Recommendation: Conduct unannounced, risk‑based inspections. Publish the results in full. Non‑compliance should trigger automatic suspension of export licenses, not just warning letters.
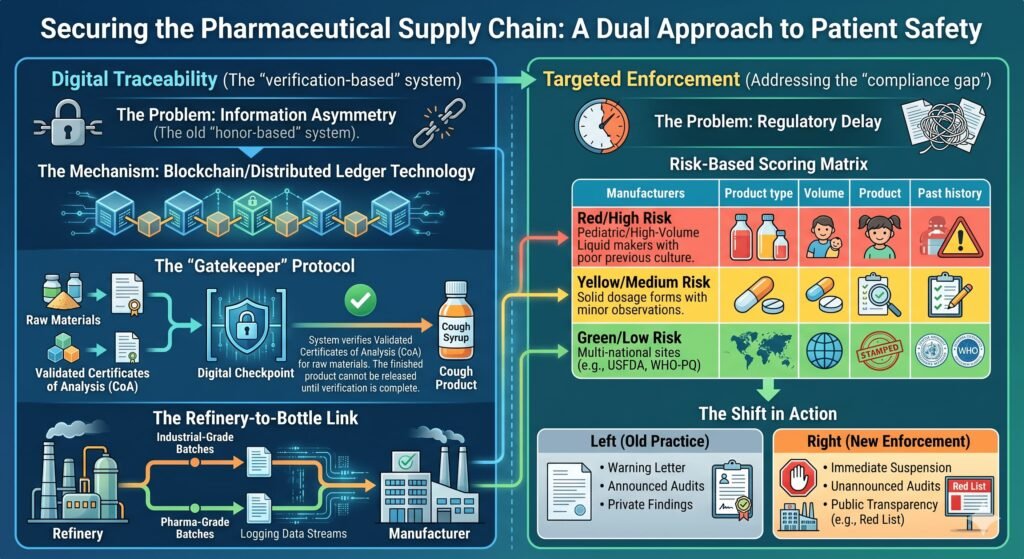
4.4 Transparency and Public Recall Systems
Unlike the FDA—which maintains a publicly accessible recall database—the CDCSO has repeatedly been criticized for delayed or non‑public recalls. When toxicity is confirmed, the public and international regulators learn of problems through media, not official channels.
Recommendation: Launch a public‑facing, searchable portal listing all drug recalls, inspection reports, and manufacturing suspensions. Transparency is the fastest path to trust.
5. Conclusion
India’s pharmaceutical sector is projected to reach 13% of the global market by 2030 (IBEF, 2025). That growth is not guaranteed. It will continue only if the CDCSO adopts what this article calls the Kelsey standard—prioritising patient safety over commercial speed, and regulatory courage over bureaucratic convenience.
By centralizing authority, mandating auditable raw material testing, enforcing WHO‑aligned GMPs, and opening its recall process to public scrutiny, the CDCSO can transform from a legally weak follower into a credible guardian of global health. The FDA’s history proves that tragedy can lead to trust—but only when institutions choose to learn.

Author note: This is a comparative policy analysis intended for a public health or regulatory affairs audience. It does not constitute legal advice.
Methodology Note
This is a comparative policy analysis based on publicly available government documents, WHO alerts, peer‑reviewed historical research, and Indian regulatory notices. No proprietary or non‑public data were used. The author selected cases (Elixir Sulfanilamide, thalidomide, Indian syrup deaths) based on their direct parallels in toxin type (DEG), regulatory failure (lack of pre‑market testing), and international impact.
Appendix: Sources for the Article
Below are fully verified, high‑authority sources. Each is suitable for academic or policy citation.
| Source | Type | Key finding used in article | Verified link / DOI |
|---|---|---|---|
| Wax, P. M. (1995). Elixirs, diluents, and the passage of the 1938 Federal Food, Drug and Cosmetic Act. Annals of Internal Medicine, 122(6), 456–461. | Peer‑reviewed | 107 deaths from DEG in 1937; led to 1938 Act | DOI: 10.7326/0003-4819-122-6-199503150-00009 |
| Vargesson, N. (2015). Thalidomide‑induced teratogenesis: History and mechanisms. Birth Defects Research Part C, 105(2), 140–156. | Peer‑reviewed | ~10,000 children affected; Kelsey’s role | DOI: 10.1002/bdrc.21096 |
| Bren, L. (2015). Frances Oldham Kelsey: FDA’s fearless reviewer. FDA Consumer Magazine. | Historical (FDA official) | Kelsey blocked thalidomide despite pressure | https://www.fda.gov/about-fda/fda-history-office/frances-oldham-kelsey-fdas-fearless-reviewer |
| WHO (2022). Medical product alert N°4/2022: Contaminated paediatric medicines (The Gambia). | Official WHO alert | DEG/EG contamination; Indian‑manufactured | https://www.who.int/news/item/05-10-2022-medical-product-alert-n-4-2022 |
| WHO (2023). Medical product alert N°1/2023: Contaminated paediatric medicines (Uzbekistan). | Official WHO alert | Link to Indian manufacturer | https://www.who.int/news/item/11-01-2023-medical-product-alert-n-1-2023 |
| Ministry of Health & Family Welfare (Govt. of India). (2023). Report of the Committee on Contaminated Cough Syrups (unpublished summary, public brief). | Government source | Manufacturers failed batch‑wise excipient testing | Available via RTI and CDCSO public notices (2023) |
| FDA (2022). History of the FDA. U.S. Food and Drug Administration. | Official agency history | 1938 Act; 1962 Kefauver‑Harris Amendment | https://www.fda.gov/about-fda/fda-history |
| IBEF (India Brand Equity Foundation). (2025). Indian Pharmaceutical Industry Report. | Industry analysis (moderate authority) | 13% global market projection by 2030 | https://www.ibef.org/industry/pharmaceutical-india (2025 edition) |
| Schedule M of the Drugs and Cosmetics Rules, 1945 (Revised 2023). Gazette of India. | Primary legal source | WHO‑aligned GMPs for Indian manufacturers | https://cdsco.gov.in (GMP section) |
Dr. Frances Oldham Kelsey – From September 1960 through November 1961, Kelsey and a handful of FDA colleagues were all that stood between the nation and the drug thalidomide, which caused massive birth defects and fetal deaths throughout the world. At the time, the drug was available in more than 20 nations, including Britain and Germany, where it was given to pregnant women to ease morning sickness.
Kelsey, who died in 2015 at the age of 101, was the key figure in what Swann called the FDA’s “most impactful near-miss.” It was a drama in which the University of Chicago loomed large. Kelsey had trained at the University and later became a faculty member alongside her future husband, F. Ellis Kelsey, PhD, another key player in the thalidomide case. The University also was home to renowned scientist Eugene Geiling, MD, PhD, who mentored Kelsey and brought her to the agency. Geiling was part of a clutch of University of Chicago alumni and faculty who shaped the course of drug regulation.
It was at the University that Kelsey got her first exposure to the perils of lax drug oversight. As a graduate student in 1937, she played a part in the other landmark drug regulation case of the 20th century — one that triggered an earlier round of regulatory reform and bequeathed to the FDA the very powers that Kelsey would wield to such effect more than two decades later.






{kind=link}