

In an earlier article in The Wire Science, I argued that India’s debate on generic medicines is routinely misframed. The issue is not whether medicines are cheap or expensive, but whether they meet defined standards of quality. Price is a market outcome; quality is a regulatory determination.
That distinction remains central, and it is worth restating because much of the current discussion continues to conflate the two.
Bioequivalence as the Foundational Pillar of Quality
As outlined previously, generic medicines are not approved merely because they resemble branded products or meet basic laboratory specifications. Their entry into the market depends on bioequivalence—evidence that the medicine behaves in the same way as the reference brand in the human body.
Regulators require manufacturers to show, through controlled studies, that key pharmacokinetic parameters fall within internationally accepted limits when compared to the branded drug. This requirement is not unique to India; it has long formed the basis of generic drug approval globally.
In regulatory terms, bioequivalence functions as the primary clinical assurance of therapeutic equivalence. Pharmaceutical equivalence—the foundational testing of assay, dissolution, purity, and stability—precedes it for the active ingredient and follows as part of manufacturing approval and ongoing batch compliance. This sequence—from laboratory standards to human biological proof—is critical and is often overlooked in public debate.

What Post-Market Testing Confirms
Recent independent testing of marketed medicines, including work that has drawn public attention in recent months, focuses on whether drugs available to patients continue to meet pharmacopoeial quality standards after approval.
The results are consistent with the regulatory framework already discussed in The Wire Science: the evidence shows no systematic difference between branded and generic medicines on these quality parameters.
Such findings do not overturn existing science; they reinforce it. Once a medicine has demonstrated bioequivalence at entry and continues to meet quality standards thereafter, price alone cannot plausibly explain large differences in therapeutic effect.
Where Responsibility for Quality Actually Lies
At this point, it is important to clarify roles.
The responsibility for certifying drug quality lies with the national drug regulator, not with individual clinicians. In India, as in other mature regulatory systems, it is the regulator—not private doctors—that determines whether a medicine is safe, effective, and of acceptable quality.
In the United States and the United Kingdom, once a medicine has been approved by the FDA or the MHRA, clinicians are expected to trust that regulatory determination rather than independently judge the drug’s manufacturing quality or bioequivalence.
Indian clinicians are similarly not expected to function as parallel quality auditors once a product has been approved by the national regulatory authority. When prescribers are implicitly asked to make quality judgments based on price or brand reputation, it reflects a gap in regulatory confidence rather than a scientific requirement.
Why Practice Has Not Followed Evidence
As noted earlier in this column, India’s move towards generic prescribing was undertaken without corresponding changes in how medicines are dispensed and explained. Doctors were encouraged to prescribe generics without routine access to bioequivalence data. Pharmacists were expected to substitute brands without being formally trained or positioned as clinical intermediaries. Patients were told generics were equivalent but were rarely shown how that equivalence is established or monitored.
In the absence of visible regulatory assurance, brands came to represent consistency—not because they were demonstrably superior, but because the system did not make quality legible to patients or practitioners.
The Substance Behind Legitimate Scepticism
Concerns about narrow therapeutic index (NTI) drugs, manufacturing consistency, and post-marketing surveillance are legitimate and have been acknowledged earlier in The Wire Science. Crucially, these concerns relate to regulatory oversight, not to price or branding. They apply to all medicines, irrespective of cost.
For NTI drugs (e.g., for epilepsy, heart conditions), the regulatory bar is rightly higher—bioequivalence limits are tightened (typically to 90-111% rather than the standard 80-125%). This underscores that the issue is one of tailored regulatory rigor and vigilant monitoring, not a fundamental flaw in the generic paradigm.
Using these valid issues to cast doubt on generics as a category diverts attention from the more substantive question of how quality is monitored, enforced, and communicated for all drugs.
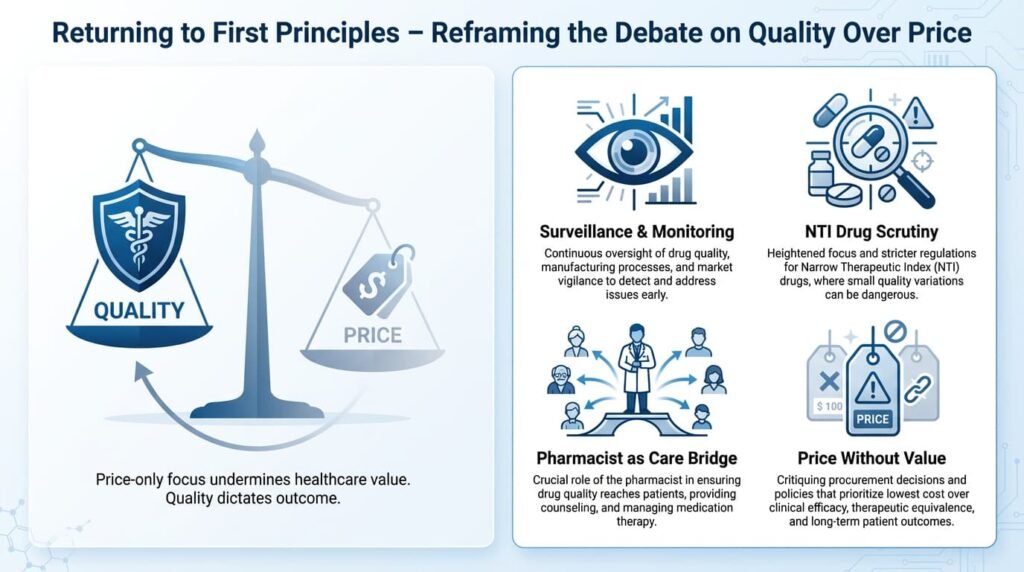
Returning to First Principles
If the debate is recentred on quality rather than price, the policy priorities become clearer—points already flagged in earlier discussions but yet to be addressed adequately:
- Public visibility of post-marketing quality surveillance: Making failure rates, inspection reports, and recall data transparent.
- Greater transparency around bioequivalence data: Especially for critical and NTI medicines, allowing informed choice.
- A clearer clinical role and accountability framework for community pharmacists: Elevating them from shopkeepers to healthcare intermediaries.
- Scrutiny of price differentials: Where no additional clinical value or proven quality premium is evident.
The Core Argument, Unchanged
Generic medicines do not derive legitimacy from being cheaper. They derive it from meeting the same scientific and regulatory standards as branded drugs—beginning with bioequivalence in human blood and continuing with quality compliance in manufacture.
Ultimately, confidence in medicines flows from confidence in regulation. If the regulator’s certification is not trusted, the solution lies in strengthening oversight, transparency, and accountability—not in outsourcing regulatory responsibility to individual clinicians or to price signals.
That is why the framing still matters.
And why it remains quality, not price.
Editor’s Note:
This article builds on an earlier The Wire Science analysis of generic medicines in India, extending the discussion to recent public debates on drug quality, bioequivalence, and regulatory responsibility.

- All Images are AI Generated for Illustration Only. E&OE
For Further Reading
This list provides resources that delve deeper into the core themes of regulatory science, bioequivalence, and public trust discussed in the article.
- On Bioequivalence & Generic Drug Science:
- U.S. FDA, “Generic Drugs: Questions & Answers” – A foundational resource explaining the rigorous approval process for generics, including bioequivalence standards.
- Davit et al., “Comparing Generic and Innovator Drugs: A Review of 12 Years of Bioequivalence Data from the United States Food and Drug Administration.” Annals of Pharmacotherapy, 2009. – A key study analyzing thousands of bioequivalence studies, concluding that generic drugs are equivalent to their brand-name counterparts.
- On Regulatory Trust and Transparency:
- Carpenter, D., Reputation and Power: Organizational Image and Pharmaceutical Regulation at the FDA. Princeton University Press, 2010. – A seminal work on how regulatory authority is built and maintained through scientific credibility and public trust.
- “The Lancet Commission on Essential Medicines Policies: Access to essential medicines: a global health challenge.” The Lancet, 2016. – Discusses the critical role of robust regulatory systems in ensuring medicine quality within access policies.
- On the Indian Pharmaceutical Context:
- Srinivasan, S., The Battle for Essential Medicines: A Citizen’s Guide to the Indian Pharmaceutical Sector. Penguin India, 2021. – Provides an accessible overview of the tensions between access, quality, and regulation in India.
- Government of India, “The Drugs and Cosmetics Act, 1940 and Rules, 1945” (Amended) – The primary legal framework governing drug quality and regulation in India. Public familiarity with its provisions is low, yet it is central to the debate.
- Reports by the Parliamentary Standing Committee on Health and Family Welfare – Various reports have highlighted gaps in India’s drug regulatory capacity and called for strengthening the Central Drugs Standard Control Organization (CDSCO).
- On Narrow Therapeutic Index (NTI) Drugs:
- U.S. FDA, “Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA” – Contains specific appendices detailing the stricter bioequivalence recommendations for NTI drugs, illustrating how science informs tailored regulation.





{kind=link}